Université Paris-Sud 11
Master (M2) de Bioinformatique et Biostatistiques 2007-2008
ARNomique et Bioinformatique de l’ARN
Séance du lundi 22 octobre 2007
A.Denise
On sait maintenant qu’aux vingt acides aminés « classiques » on peut en ajouter deux : la Sélénocystéine et la Pyrrolysine. La pyrrolysine, le « 22ème acide aminé », est présente dans des gènes de certaines archae méthanogènes. Une particularité de cet acide aminé est que son codon est aussi un codon stop : UAG. Dans les CDS (Coding DNA sequences) considérés, il y a donc un codon stop UAG en phase. Au cours de la traduction, ce codon peut soit être considéré comme un codon stop « normal » par la machinerie de traduction, auquel cas la traduction s’arrête là, soit considéré comme codant pour une pyrrolysine, auquel cas celle-ci est incorporée dans le peptide en cours de construction et la traduction continue jusqu’au stop suivant.
Selon certains auteurs, une structure secondaire particulière en 3’ du codon UAG favoriserait, dans ces archae, l’incorporation de la pyrrolysine[1]. Une structure en épingle à cheveux a été déterminée en combinant des expériences de probing et des alignements manuels des sites considérés. La figure ci-dessous résume les résultats du probing.
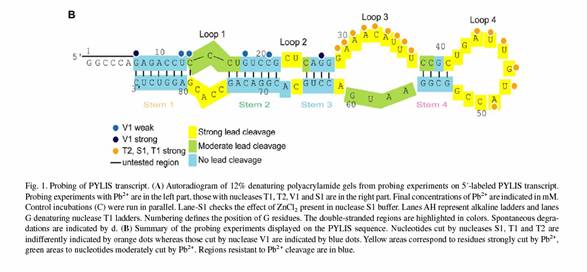
La structure qu’ils proposent, commune à au moins six CDS de trois organismes différents, est la suivante :
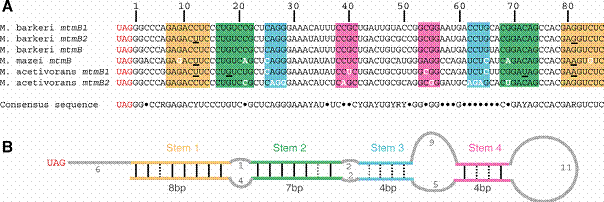
Votre travail va consister notamment à tenter de retrouver cette structure[2] en utilisant exclusivement des outils bioinformatiques. On notera aussi que vous ne travaillerez pas vraiment en conditions réelles: en général, on ne connaît pas à l’avance la structure que l’on recherche !
Les gènes connus jusqu’ici pour être soumis à l’incorporation d’une pyrrolysine sont disponibles dans une base de données en ligne : http://www.lri.fr/~zhouyu/pylis/. Sélectionnez dans la base les séquences des gènes mtmB ; demandez les séquences « readthrough », pour n’obtenir que la partie de la séquence à partir du codon UAG en question. Copiez les cinq séquences correspondant à celles de la figure ci-dessus et sauvegardez-les dans un fichier[3]. (La séquence de M. Barkeri mtmB est exactement la même que celle de M. barkeri mtmB1, c’est pourquoi il est inutile de la considérer.)
Tout au long du travail, vous pouvez utiliser VARNA, une applet Java téléchargeable à http://www.lri.fr/~ponty/VARNA, pour visualiser les structures secondaires que vous obtiendrez.
- Prédiction
comparative de structure, phase 1.
Allez sur le site de CARNAC : http://bioinfo.lifl.fr/RNA/carnac/.
Lisez la présentation, puis choisissez la version en ligne (web server).
Lancez l’application sur l’ensemble des cinq séquences. Dans la fenêtre
des résultats, choisir « Visualize all foldings with
RNAfamily ». Vous remarquez des éléments de structure communs dans
les cent premiers nucléotides (environ) en aval de l’UAG.
- Prédiction
ab initio. Allez sur le serveur de mfold (http://frontend.bioinfo.rpi.edu/applications/mfold/).Tronquez
les cinq séquences pour ne garder que la zone intéressante trouvée
précédemment et repliez-les avec mfold. Comparez les résultats,
visuellement, à la structure recherchée.
- Comparaison de structures. Pour comparer
plus objectivement les structures obtenues à la structure recherchée,
utilisez RNAForester. (http://bibiserv.techfak.uni-bielefeld.de/rnaforester/submission.html)
ou bien migal (http://igm.univ-mlv.fr/~allali/migal/index.php).
- Prédiction
comparative, le retour. Avec les séquences tronquées, essayez à nouveau
CARNAC, puis RNAshapes (http://bibiserv.techfak.uni-bielefeld.de/rnashapes/submission.html)
puis RNAalifold (http://rna.tbi.univie.ac.at/cgi-bin/alifold.cgi),
en faisant varier les paramètres et l’ensemble des séquences (par exemple,
supprimez une ou plusieurs séquences de l’ensemble). Notez que RNAalifold
requiert que les séquences soient préalablement alignées.
- Prédiction
comparative, fin. Remplacez les deux séquences de M. acetivorans par celles –ci :
>Chimere1
UAGGGCCCAGAGACCUCCGAGUGACCUGACCGAAAUAUUUGGACUGACUGCGCCGGUCCAAUGAGGUCCAGUCACUCCCACGAGGUCUCGCAACUAAACG
>Chimere2
UAGGGCCCAGAGACCUCCGUGUGAGCUGAGCGAAAUAUUUCCACUGACUGCGCCGGUGGAAUGAGCUCCACUCACACCCACGAGGUCUCGCAACUAAACG
et relancez les programmes de la question précédente. Que remarquez-vous ? Comment l’expliquez-vous ?
- Modélisation
du site PYLIS. Nous supposons maintenant que la structure (que nous
appellerons dorénavant le site PYLIS)
est connue et avérée. Il s’agit de modéliser ce site et de le décrire afin de pouvoir le rechercher avec un
programme tel que Darn! (http://carlit.toulouse.inra.fr/Darn/).
Lisez la documentation de Darn! et écrivez un descripteur qui décrit les
cinq structures (ou le plus possible d’entre elles). Vérifiez avec que ces
structures sont bien trouvées par Darn! dans les CDS considérés.
- Evaluation
de la spécificité. En utilisant GenRGenS (http://www.lri.fr/~genrgens/)
engendrez des séquences aléatoires de composition similaire à celles des
CDS considérés (en Bernoulli et/ou en Markov d’ordre 1), recherchez les
sites PYLIS avec Darn! et évaluez le taux de faux
positifs obtenu.
- Sites PYLIS aléatoires. GenRGenS est capable d’engendrer des séquences structurées, en utilisant des grammaires non contextuelles. Lisez la documentation (http://www.lri.fr/~genrgens/manual/GRGs-manual-html) et inspirez-vous notamment de l’exemple donné en http://www.lri.fr/~genrgens/manual/GRGs-manual-html/node5.html#SECTION00543000000000000000 pour engendrer des séquences dont la structure prédite est proche de celle du site PYLIS.