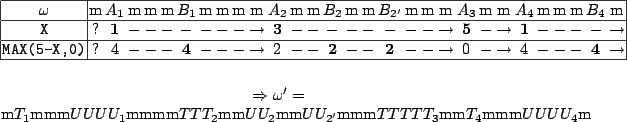Sometimes, true uniformity or total control over the distribution
of the sequences is not needed, and the hierarchical aspects of
some models can be captured. The MASTER generation is then a good
tradeoff between computational efficiency and expressivity.
The main principle is to generalize the idea behind hidden Markovian
to any type of master and hidden states models.
At first, a sequence is derived from a master description file, having
a length passed to the main generation engine of GenRGenS. Then each
of its symbols can either or not be defined non-terminal and
rewritten using another description file coupled with a size distribution.
This class of models has been included to allow different models to be combined. For example, a simple model for the
Intergenic/ORF alternation could describe the framed aspect of the
ORFs (using the Framed Markov model) as well as the lack of a start codon in the
intergenic areas (using the Rational expression model). This can be modelled using our hierarchical models.
Hierarchical models comprise a main master model, a set of auxiliary models and a set of rules
defining how to rewrite letters of the master sequence using the auxiliary models. This can also be seen as a generalisation of the HMMs,
as the hidden state sequence can be computed independently from the letters
arising from each hidden state.
A sequence length distribution can be provided with each rule through an expressive language, enabling complex constraints such as
the overall length of the sequence is a multiple of to be expressed.
It is noticeable that this approach to random can save some time and
space while using models that require more than linear time and space
complexities. Indeed, if and are the sizes of the two parts
of an expected sequence then
, for
all
.
It also implies a small loss of control over the distribution of
sequences. For instance, a model built up by concatenating two
sequences chosen uniformly is unlikely to be uniform itself.
More generally, let and be two models, a sequence issued
from the model may admit different decompositions
.
Its global probability is then
,
which induces a bias if uniformity is aimed at.
Furthermore, as it is impossible to constrain the ending of a Markov
chain, the concatenation of two sequences issued from some Markovian
models may induce some bias at the cutpoint.
Optional, defaults to SYMBOLS = WORDS
Chooses the type of symbols to be used for random generation.
When WORDS is selected, each pair of subsequent symbols is separated with spaces during the
generation.
Required
Defines the main random generation model description file. The file must be accessible by appending the
string
to the current directory.
A so-called master sequence is generated from this description file for further rewritings.
Its length equals to the size provided to GenRGenS.
Remark: The overall size of the sequence generated from a MASTER model is usually not related
to the size parameter provided to GenRGenS.
Required
Defines the way to expand each occurrences of a symbol from the main file to sequences issued from ,
having size given by the distribution .
Each is a symbol that must belong to the vocabulary of the master file, but not necessarily to the
sequence generated from it.
is the path to a description file that must be read-access enabled.
is a description of the random value associated to the size of this symbol's expansion.
A size can either be a constant, one of the predefined random variate generator,
or an arithmetic expression defined recursively.
-
integer or float constant: Generates a constant sequence size. If a non-integer size is specified at top-level,
the value is rounded to the closest integer value, as it would be have been using round.
-
+,-,*,/,^ : Classical binary arithmetic operators, acting on real numbers
and returning a real value.
-
(): An expression nested by parenthesis is evaluated separately and priorly from its context.
It can be used to overrule usual operator precedences. Ex : +*(+)*
-
pow(,): Functional form of the power operator ^ , returns . If first argument is
omitted, returns .
-
log(,): Returns
. If is omitted, returns
.
-
floor(): Returns the closest integer value smaller than .
-
round(): Returns the closest integer to .
-
ceil(): Returns the closest integer value greater than .
-
min(,): Returns the smallest number among {}.
-
max(,): Returns the smallest number among {}.
-
length: Returns the former length of the master sequence. Equivalent syntax: size or N.
-
normal(,): Generates a pseudorandom real value obeying a normal
law having mean and standard deviation . Equivalent syntax: gaussian(,).
-
uniform(,): Generates a pseudorandom integer value uniformly distributed over (e.g.
).
If is omitted, defaults to 0. Equivalent syntax: random(,).
-
var(,): Declares a variable , whose value always equals the most recent evaluation of .
Once declared, can be used anywhere among the SIZE declaration parts of the ggd file. Each variable carries the value 0 as
long as it is not assigned.
MASTER package introduces a way to capture the dependency between two subsequences' size.
A typical dependency that one may find useful would be : The total size of the two subsequence
is 100nt, although the first's size has been observed to vary with respect to a given distribution.
The MASTER package offers the possibility to declare variables, which is a powerful and
delicate feature.
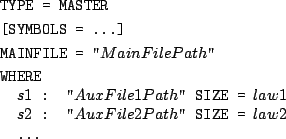
Inside of a MASTER description file, simply enclose any expression of a length definition inside
a VAR declaration. Ex.:
TYPE = MASTER
MAINFILE = "MainFile.ggd"
WHERE
A : "AuxFile1.ggd" SIZE VAR(X,UNIFORM(0,5)+1)
B : "AuxFile2.ggd" SIZE MAX(5-X,0)
In this example, each evaluation of the random variable UNIFORM(0,5)+1 is assigned to a variable named X.
Each evaluation of the expression MAX(5-X,0) will then make use of the current value of X. This is likely
to trick one into bad assumptions over the resulting sequences, for instance if the sequences issued from MainFile.ggd
do not only present simple A-B alternations. Suppose that MainFile.ggd contains an implementation of a simple
RATIONAL model, based on the regular expression (A|B|m)*, that AuxFile1.ggd and AuxFile2.ggd are
RATIONAL models based on the regular expressions T* and U*, and that a sequence is issued
from MainFile.ggd. Rewritings according to the SIZE definitions above result in the following sequence .
Figure 6.2:
Dependencies during generation stage
It can be seen in figure 6.2 that a variable's assignment can either be used by several subsequent
dependant variables assignments, as in
, or be ignored like .
Therefore, this feature should only be used when the sequences
issued from MainFile.ggd are sufficiently constrained, for instance by showing a strict assignment/use alternation such
as the regular expression m*(Am*Bm*)*.
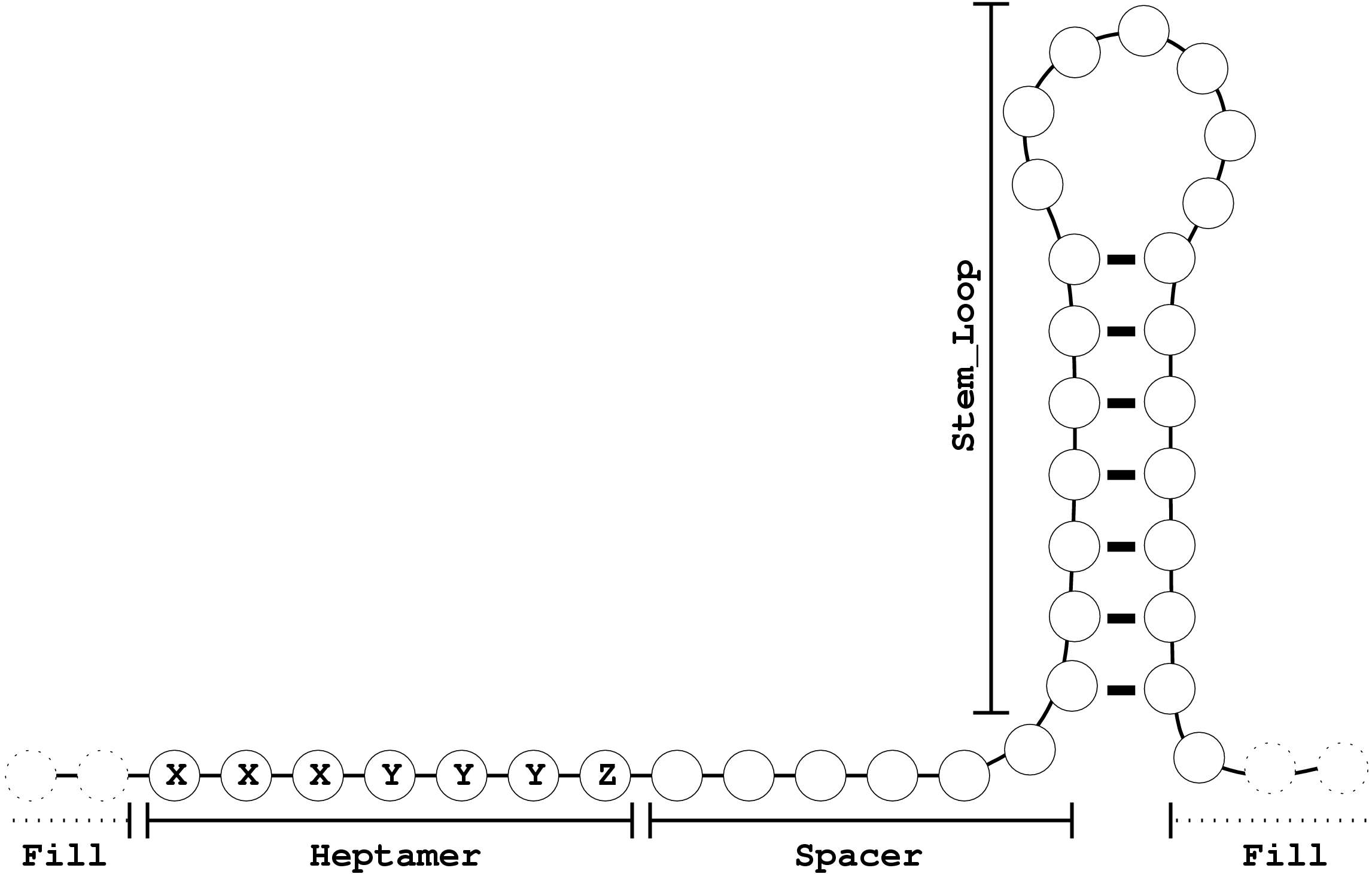
This example is inspired by a simple model for stem-loop ribosomal -1 frameshifting sites. A frameshifting RNA causes the ribosome to
slip over a specific sequence/structure motif and shift to another phase. A model inspired by the HIV-1 stem-loop
frameshift site is detailed in figure 6.3.
Figure 6.3:
A simple stem-loop frameshift model. X stands for any base,
Y means AorU and Z is anything butC
The Heptamer part can be modeled with a regular expression, similar to a mutation model. The Spacer will use a Bernoulli model(model of order 0). The
Stem_Loop requires the full power of a context free grammar. Lastly, the frame shift sites are drowned into an ocean of coding
RNA(symbol Fill) modeled again by a markov model.
Figure:
A MASTER description file for the frameshift model from figure 6.3.1
On line 2, we use words as symbols to avoid confusion.
On line 3, we define the main description file, whose content is listed in figure 6.5.
On line 5, we ask the generator to rewrite each occurrence of the symbol Heptamer with a sequence
issued from heptamers.ggd, having size .
On line 6, we ask the generator to rewrite each occurrence of Spacer with a sequence
issued from spacers.ggd, using a size uniformly distributed over . Note that
UNIFORM(5,8) is an equivalent syntax for 5 + UNIFORM(0,3). After each evaluation,
the resulting size is stored inside a variable X.
On line 7, we ask the generator to rewrite each occurrence of Stem_Loop with a sequence
issued from stem_loops.ggd, using a size uniformly distributed over . After each evaluation,
the resulting size is stored inside a variable Y.
On line 8, we ask the generator to rewrite each occurrence of Fill using a size that depends on
the previous assignments.
The somehow cabalistic formula for the size of Fill's expansion is just a little trick to ensure that
the Heptamer motif always occurs on phase 0. That is, NORMAL(300 , 100)+X+Y+7 must be a multiple
of . So, by dividing by and rounding to the closest smaller integer and then multiplying by , we get the closest
sum of the sizes of Heptamer, Spacer, Stem_Loop and Fill that is divided by without
a remainder. It suffices then to substract 7 (Heptamer's size), X (Spacer's size) and
Y (Stem_Loop's size), to get an eligible size for Fill.
Next:Bibliography Up:GenRGenS v2.0 User Manual Previous:The RATIONAL package :Contents
Yann Ponty
2007-04-19